今回はネット上のサービスであるSignalPを使って、目的タンパク質にシグナルペプチドが含まれるか予測します。
SignalP-5.0のURLはこちら
→http://www.cbs.dtu.dk/services/SignalP/
ちなみにSignalP-4.1やSignalP-3.0などもありますが、SignalP-5.0だとシグナルペプチドの候補だけを出力できるので便利です。
今回はSignalPの使い方だけでなく、ファイル処理の方法も紹介しています。
タンパク質の候補を得る
今回はモデル植物のシロイヌナズナ(Arabidopsis thaliana)を例に進めます。
はじめに調べたいタンパク質の配列を取得します。
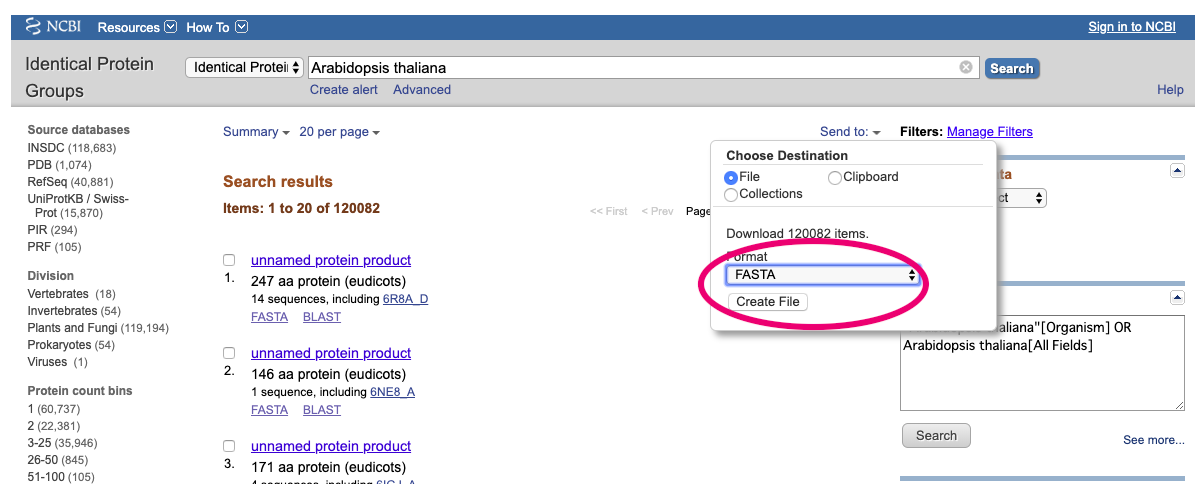
Arabidopsis thalianaをIdentical Proteinで検索すると一覧が出てきます。
Send to→File→カーソルからFASTA形式を選び→Create Fileでダウンロードします。
しばらくすると、sequence.fastaというファイルがダウンロードされます。
早速タンパク質の情報を手に入れたので、SignalPに行きたいところですが…
SignalPでは1ファイル当たり、5000個のタンパク質しか解析できません。
今回のファイルは120000以上のタンパク質が含まれているので、そのままでは解析できません。
そこで、ファイルを分割します。
Macであれば、ターミナルというソフトがあるので、コードを打ち込むと分割したファイルを作ってくれます。
Linuxでは端末、Windowsではコマンドプロンプトがターミナルに相当します。
仮にsequence.fastaがデスクトップのtestフォルダに入っているとします。
始めにターミナルを開きます。
開いたら下のコードを打ち込み、enterを押します。
Mac, Linux
cd Desktop/testWindows
CD Desktop/testすると、今はDesktopにあるtestフォルダ内で作業しますよ〜という状態になります。
Linuxでも同様にcdコマンド、WindowsではCDコマンドになります。
次に下のコードを打ちます。
こちらのコードだと2000行ずつ、ファイルを分割することができます。
Mac, Linux
split -l 2000 sequence.fasta sequence_目的の数字に変えると、その行数のファイルを作ることができます。
上のコードを使うとsequence.fastaがsequence_aa、sequence_ab、sequence_ac…と分けられます。
ただ問題もあり、それぞれのファイルはタンパク質ごとに分割されたわけではありません。
aaのファイルだと、最後は配列の途中で切れている。
abは最初と最後が配列の途中で切れている。
ということが起きますので、調整しなければなりません。
ですが、行単位で切れるため後処理が楽です。
Mac, Linux
split -b2m sequence.fasta sequence_これはsequence.fastaを2MBごとに分割し、sequemce_**というテキストで出力するという意味です。
2mの数字を変えれば、目的の大きさのファイルにすることができます。
Windowsの場合、Makecabコマンドになるようですが、残念ながら僕には難しかったです…
Makecabで調べてみてください。
今回のファイルは約60MGだったので、30つのファイル(sequence_aa, sequence_ab, sequence_ac…)に分割されました。
こちらも同様にファイルの最初と最後で配列が切れてしまう問題があります
もし、綺麗に分けれる方法を知っていたら教えてください。とても助かります。
SignalPで解析する
次にSignalPのページに行きます。
赤丸で囲っているUpload Fasta Fileというボタンを押し、先ほどのファイルをアップロードします。
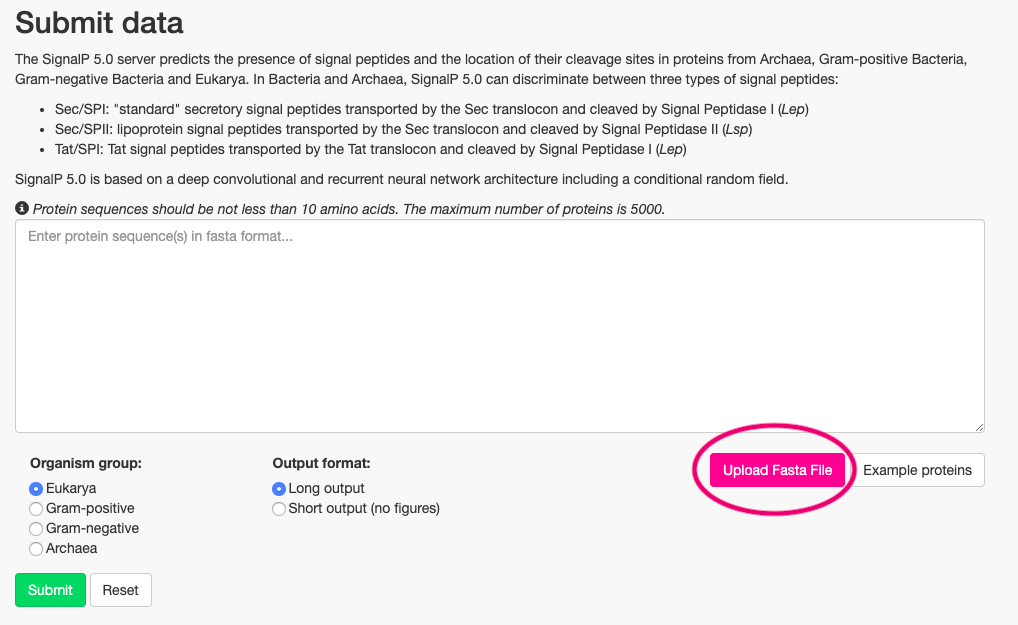
配列をコピーして、直接白いボックスに貼り付けてもできます。
今回は真核生物なので、Eukaryaを選択し、アウトプットの形式はLong outputを選びます。
緑のボタンのsubmitを押すと解析が始まります。
ファイルサイズが大きければ大きいほど解析に時間がかかります。
僕の環境ですと1ファイルの大きさが約70Bで、解析に1分程度かかります。
可能な限り小さい方が解析は早くなります。
ですが、出力されるときは元のファイル名は引き継がれず、解析結果も一つ一つ出力する必要があるため、混乱しやすいです。
解析後、結果を確認
解析が終わるとこのような画面になり、解析されたタンパク質の数が表示されます。
今回は解析時間短縮のため、小さいファイルを使ったため149と少ないです。
Downloadのタブをクリックすると、いくつか出力の形式があります。
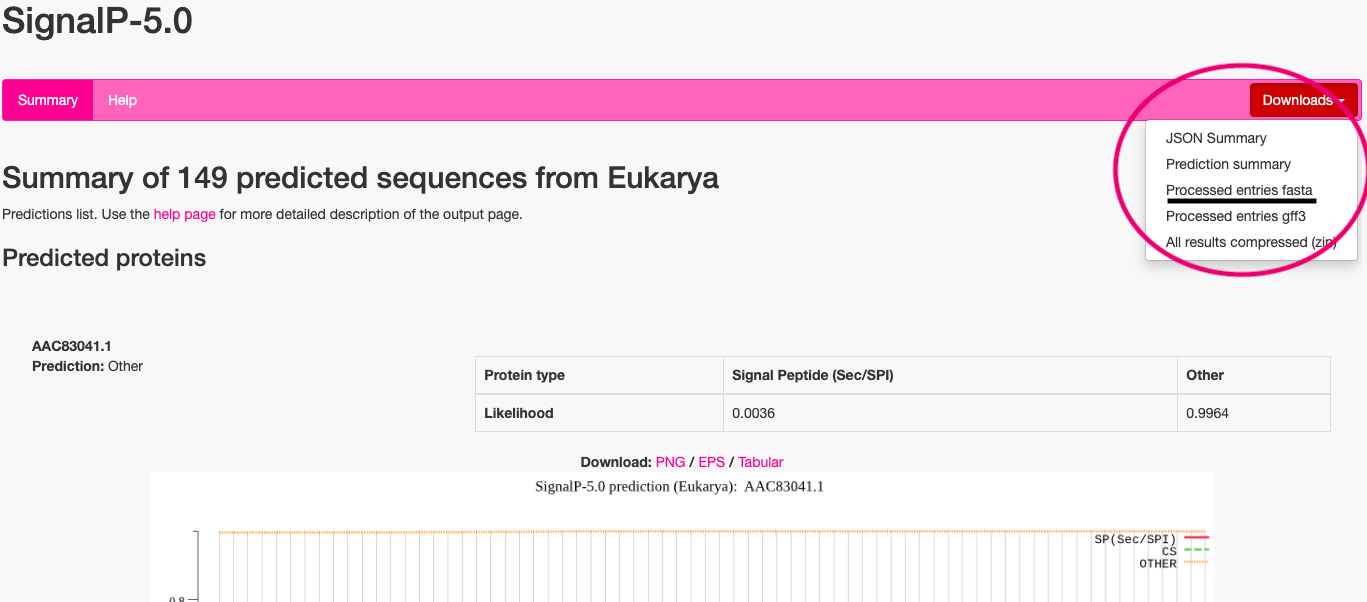
Processed entries fastaを選ぶと、シグナルペプチドと予測されたタンパク質のみをoutput_mature.fastaというファイルで出力します。
その他のタンパク質も含んだ結果はPrediction summaryに、グラフのpngファイルも含んだ結果は.zipで出力できます。
今回はProcessed entries fastaのみ確認します。
解析するとfastaファイルがたくさん出力されるので、それぞれを一つのファイルに統合します。
前回と同様に始めにターミナルを開き、fastaファイルが入っている場所までcd(CD)コマンドで移動します。
次に統合するために以下のコードを打ちます。
Mac, Linux
cat *.fasta >SignalP_result.txtWindows
type *.fasta >SignalP_result.txtLinuxでも同じcatコマンド、Windowsではtypeコマンドです。
cat/typeで.fastaで出力された全てのファイルをSignalP_result.txtという一つのファイルに出力します。
*は.fastaのファイル全てという指定です。
ちなみに以下のコマンドで行番号も付与することができます。
Windowsのコマンドは違うようなので、分からないです。
Mac, Linux
cat -b *.fasta >SignalP_result.txtですが、アクセッション番号と配列の行の前に番号が振られてしまいます。
そのため、最後の番号を割る2すると、予測されたタンパク質の数が分かります。
これでSignalPでの作業が終了です。
終わりに
今回はSignalPの使い方をまとめました。
これでシグナルペプチドと予測されるタンパク質を絞ることができます。
ですが、SignalP-5.0のホームページには以下の文章があります。
“Remember, the presence or absence of a signal peptide is not the whole story about the localization of a protein! If you want to find out more about the sorting of your eukaryotic proteins, try the protein subcellular localization predictor.”
あくまで解析の結果は予測であり、シグナルペプチドであることを完全に担保することはできません。
詳細の解析も行うようにしましょう。
ちなみにtogotvでもSignalPの使い方が紹介されているので貼ります。
こちらは古いバージョンのSignalPについてですが、ちゃんとグラフの説明もこちらではしています。

コメント